Malattia Gaucher, diagnosi precoce per tornare a vita “normale”
11 Ottobre 2016 - di Mari
GENOVA – Un ragazzino di 11 anni che, dopo la diagnosi di Malattia di Gaucher e l’inizio della terapia, ha passato la sua prima estate da bambino “normale”. Una ragazzina alla quale, dopo diversi anni senza una diagnosi, ne è stata riconosciuta una e ora è sotto cura. E ancora, i progressi nella terapia, con nuove potenzialità, tra le quali quella per bocca con eliglustat, che si affianca alla terapia per infusione, o la sperimentazione di un farmaco che dà buone speranze ai pazienti affetti dalla Malattia di Gaucher di tipo 3, che sono il 5% del totale degli affetti dalla patologia e che, finora, non avevano a disposizione una terapia efficace.
Sono alcune delle buone notizie che arrivano dal XII Incontro sulla Malattia di Gaucher, che si è tenuto martedì 11 ottobre al Palazzo della Borsa di Genova, dove un centinaio di medici e specialisti italiani e stranieri si sono confrontati su questa patologia rara.
I messaggi che la comunità di scienziati vuole inviare sono che la malattia si può diagnosticare in età precoce, che si può trattare, che ci sono nuove prospettive di trattamento e che gli studi, compresi quelli italiani, hanno avuto e stanno avendo degli esiti positivi. Tutto questo per permettere ai malati, che sono 10.000 circa nel mondo e qualche centinaia in Italia, di vivere una vita quanto più possibile normale.
Vista la rarità della patologia, che nei due terzi dei casi ha appunto un esordio in età pediatrica, tra i medici c’è chi ha anche solo un paziente affetto dalla Malattia di Gaucher, ma ciò non toglie che, ogni volta, gli esperti si impegnino per essere presenti all’incontro, tutti guidati da un concetto: le terapie, come quella enzimatica sostitutiva e quella innovativa orale con eliglustat, per esempio, sono efficaci se iniziate precocemente e possono evitare complicanze irreversibili.
Tra i professori al Convegno di Genova c’è anche il professor Timothy M. Cox, docente all’Università di Cambridge, uno dei principal investigators sulla patologia, che porta i risultati della terapia del substrato. Il dottor Seng H. Cheng racconta invece le nuove terapie e la loro efficacia nelle patologie correlate, il pediatra Nicola Tovaglieri del Niguarda di Milano, invece, spiega come è riuscito a diagnosticare la malattia in una ragazzina. Mirella Alpa, nefrologa reumatologa e immunologa al San Giovanni Bosco di Torino parla del lavoro riguardo alla patologia, Maria Domenica Cappellini, docente all’Università degli Studi di Milano tratta il tema del metabolismo del ferro nei pazienti affetti da Malattia di Gaucher di tipo I. E ancora, Alberto B Burlina, direttore dell’Unità Operativa Complessa di Malattie Metaboliche Ereditarie dell’Azienda Ospedaliera di Padova, quello dello screening neonatale per la Malattia di Niemann Pick. Irene Motta, ematologa all’Università di Milano, nel suo intervento, sottolinea che “Non è mai troppo ta rdi per la genetica”. Francesca Carubbi e Fabio Nascimbeni docenti al Dipartimento di Scienze Biomediche, Metaboliche e Neuroscienze dell’Università di Modena e Reggio approfondiscono l’argomento delle modifiche metaboliche e l’incidenza della Sindrome metabolica nella Malattia di Gaucher nel fegato.
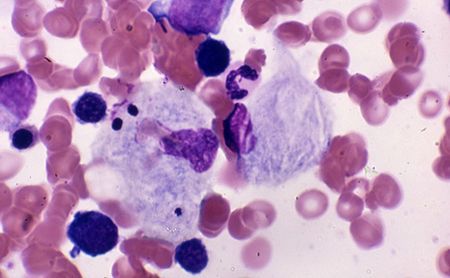
La Malattia di Gaucher è una condizione rara ed ereditaria con una frequenza intorno a 1 su 40.000 persone. A causa della carenza di un enzima, la beta-glucosidasi acida (glucocerebrosidasi), che ha il ruolo di scindere una molecola di natura lipidica, la malattia porta un accumulo di lipidi nelle cellule denominate appunto di Gaucher. Questo provoca l’ingrossamento della milza e del fegato con anemia, ematomi, sanguinamento eccessivo, ma anche osteoporosi o alterazioni ossee.
La malattia di Gaucher fu descritta per la prima volta nel 1882, ma gli studi clinici sono partiti molti anni dopo. Basti pensare che solamente nel 1984, grazie alla madre di un bambino statunitense e a un gruppo di ricercatori, si iniziò la sperimentazione della terapia con l’enzima mancante.